CASE REPORT | https://doi.org/10.5005/jp-journals-10001-1346 |
Surgical Management of Unilateral Craniosynostosis: A Case Report
1–4Department of Dentistry, SKIMS Medical College and Hospital, Srinagar, Jammu and Kashmir, India
Corresponding Author: Munaza Shafi, Department of Dentistry, SKIMS Medical College and Hospital, Srinagar, Jammu and Kashmir, India, e-mail: drmunaza.shafi@gmail.com
How to cite this article Shafi M, Shakeel M, Ahad M, et al. Surgical Management of Unilateral Craniosynostosis: A Case Report. Int J Head Neck Surg 2020;11(2):39–41.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Craniosynostosis is the premature fusion of cranial sutures which can result in neurological abnormalities, facial and skull deformations, and increased intracranial pressure. It can be diagnosed by physical examination, the use of plain film X-rays, computed tomography (CT) exams, or with the use of ultrasound. If caught within the first few months of life, or prenatally, craniosynostosis can be treated endoscopically with minimal invasiveness. Those cases that are discovered after the first 6 months of life can be treated surgically with cranial vault remodeling. Both procedures have an excellent success rate and a low recurrence rate. In this case report, a 6-month-old healthy boy with deformity of the left forehead and orbit that is caused by frontal plagiocephaly and coronal unilateral synostosis is presented. This abnormality was corrected by frontal craniotomy and fronto-orbital complex advancement under general anesthesia.
Keywords: Fronto-orbital advancement, Plagiocephaly, Unilateral craniosynostosis.
INTRODUCTION
Craniosynostosis is premature fusion of one or several sutures in the skull bone.1 Premature fusion of one of the coronal sutures results in plagiocephaly (oblique skull) occurring in 1 in 10,000 live births.2 Frontal plagiocephaly is caused by deformational or synostotic forces.2–4 Growth of the brain is continually stimulating the bones in the skull to expand and create new bone at the suture lines. This continues to happen until around the fourth decade of life but is most valuable in the first year when brain growth quadruples. The normal range for infant fontanelles to fuse is from 4 to 26 months, with semipermanent fusing by the age of 12 and permanent fusing occurring by age 40 or 50.5 Craniosynostosis is usually present at birth but may not be diagnosed until the early months of the child’s life.5
The signs of unilateral coronal synostosis include a flat frontal bone, retardation of the supra and lateral orbital rim, elevation of the sphenoid wing producing a harlequin appearance on plain radiography or computed tomography (CT) scan, and deviation of the root of the nose toward the affected side.1,3,6,7
The cause of craniosynostosis for the most part is unknown. Cases can be classified as either primary, which is an intrinsic defect in suture, or secondary, which is premature closure of normal sutures because of another medical condition such as deficient brain growth.8 Primary craniosynostosis, from an etiologic view, can either be idiopathic or familial.8 Familial cases usually present as a type of craniofacial syndrome and can be the result of agenetic mutation.8 These known mutations happen in the fibroblast growth factor receptors, which modulate cell proliferation, differentiation, and migration.8 The mutations are associated with Pfeiffer, Apert, Crouzon, Beare-Stevenson, Jackson-Weiss, and Muenke syndromes.8 Secondary craniosynostosis is developed from a known underlying disorder. These disorders can be systemic and metabolic and include conditions such as “hyperthyroidism, hypercalcemia, hypophosphatasia, vitamin D deficiency, renal osteodystrophy, Hurler’s syndrome, sickle cell disease, and thalassemia.”8 In this case report, we introduce a patient with nondeformational unilateral plagiocephaly.
CASE DESCRIPTION
A 5-month-old infant with left-sided plagiocephaly was referred to our department. Examination revealed a healthy boy infant who was normal apart from the craniofacial deformity. The mother had a normal pregnancy and delivery and she did not use any medication. Physical examination showed an oblique head. The left ipsilateral forehead and parietal were flat and retruded. The left temporal bone was depressed. The left supraorbital ridge and lateral orbital rim were depressed and displaced posteriorly. The right side revealed frontal and parietal bossing (Fig. 1). CT revealed a unilateral coronal synostosis (harlequin appearance) (Fig. 2).
A bicoronal incision was made, and Raney clips were placed on both sides of the wound to maintain meticulous hemostasis. The scalp was dissected to the temporal and supraorbital region through the subgaleal plane (Fig. 3). At the upper border of the temporalis muscle, the incision was made through the periosteum and the muscle was left attached to the scalp. Deep to the temporalis muscle, the temporalis fossa was exposed. After fronto-orbital advancement, the bone parts were fixed with titanium screw and miniplates by the maxillofacial surgeon (Fig. 4). Osteotomy and the additional bone cuts in the frontotemporal region created enough space for expansion of the brain. The fragments of the bone were inserted between the globe and the brain. After completion of this surgery, the patient was put on follow-ups on neurosurgeon, maxillofacial surgeon, and ophthalmologists for regular check-up.

Fig. 1: Pretreatment picture

Fig. 2: 3D CT showing unilateral craniosynostosis
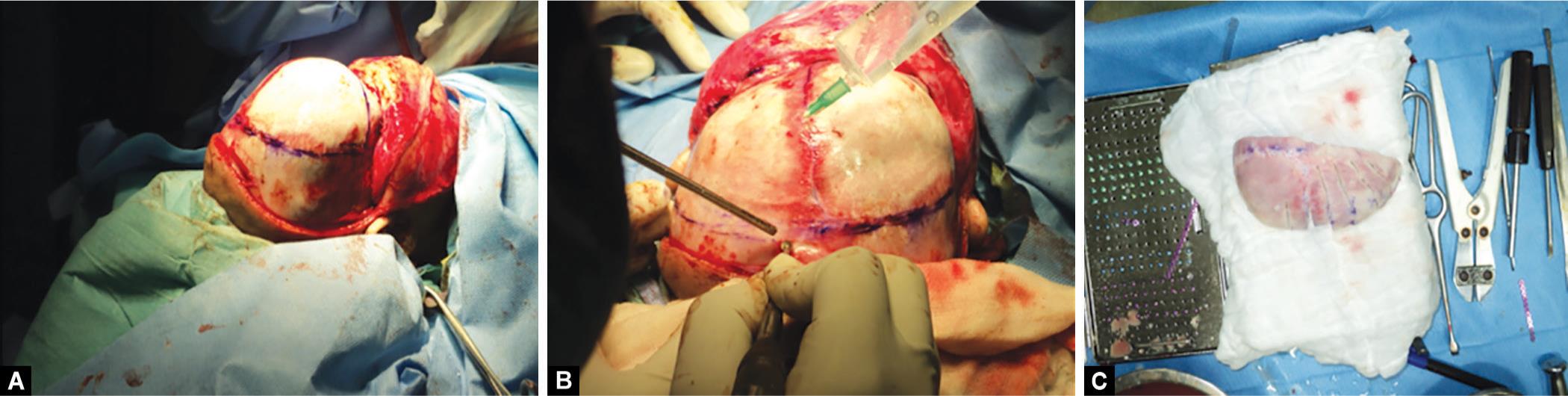
Figs 3A to C: Dissection of scalp. Remodeling of frontal bone to allow the growth of brain
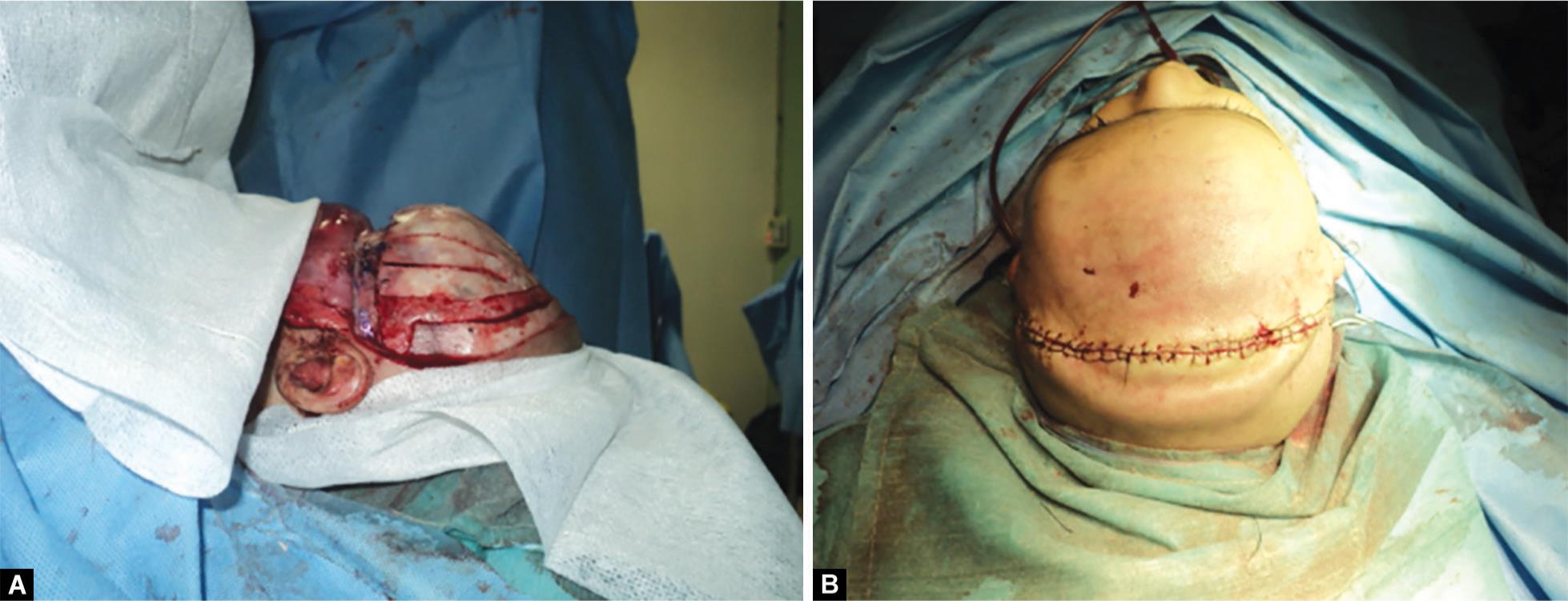
Figs 4A and B: Fronto-orbital advancement. Scalp sutured back in place done with plates in place
DISCUSSION
Craniosynostosis is premature closure of one or multiple cranial sutures. The sagittal suture is affected in 40 to 60 percent of cases, the coronal suture in 20–30% of cases, and the metopic suture in less than 10% of cases; true lambdoid synostosis is rare.9 Suture and fontanelle closure takes place in different times.10 For example, range of normal closure of the anterior fontanel is 4–26 months and posterior fontanel is birth to 2 months. Cranial development and mature sutural closure occurs by the age of 12, while the completion of sutural fusion by ossification is an ongoing process until the third or fourth decade of life.10,11 Of affected individuals, 2–8% have primary craniosynostosis. Secondary causes include rickets, hyperthyroidism, hypercalcemia, bone marrow hyperplasia, or inadequate brain growth (microcephaly and shunted hydrocephalus). Usually, craniosynostosis is present at birth, but it is not always diagnosed when mild. Generally, it is diagnosed as a cranial deformity in the first few months of life.
Clinically in positional plagiocephaly, the skull has a rhomboid form, and in synostotic plagiocephaly, it is trapezoidal.12 Retrospective studies have shown that the basilar coronal ring is involved in one-third of the patients with unilateral coronal craniosynostosis.12,13 Frontosphenoidal synostosis is a rare cause of frontal plagiocephaly. Unilateral coronal synostosis accounts for 20 to 30% of synostoses in infancy. Most cases of unilateral coronal synostosis are sporadic and nonsyndromic. There are, however, examples of familial UCS, such as Seatre-Chotzen syndrome.14 Unilateral coronal craniosynostosis is the most common hereditary form of single-suture fusion. First described in 1994, several families with hereditary “nonsyndromic craniosynostosis” have been reported with a variable occurrence of unilateral or bilateral coronal synostosis.15 In 1997, a mutation in fibroblast growth factor receptor 3 (FGFR3P250R) was described in patients with apparently isolated coronal craniosynostosis and is now known as Muenke syndrome.16 This FGFR3P250R mutation has since been identified in approximately 10% of isolated unilateral coronal synostosis and a greater percentage of bilateral cases.16
Unicoronal synostosis causes regional growth restriction and compensatory expansion of the neighboring tissues, producing overt fronto-orbital dysmorphology. Infants with synostotic frontal plagiocephaly (unilateral coronalsynostosis) have a characteristically asymmetric face and anterior cranium. Characteristic deformities ipsilateral to the synostosis include flattening of the frontal bone and ipsilateral forehead, ipsilateral elevation–recession of the supraorbital rim, and narrowing and lateral deviation of the orbit. The nasal root is deviated toward the affected side, whereas the cartilaginous framework is rotated slightly to the contralateral side, as is the chin point, and elevation of the ipsilateral ear. This midfacial rotation also accounts for the anterior auricular position on the affected side. On the affected side, palpebral fissure is rounded and shortened transversely. On the contralateral side, the forehead is bossed, the supraorbital rim is rotated anteroinferiorly, and the palpebral fissure can be slightly narrowed.17 Bruneteau and Mulliken believe that physical examination focusing on the supraorbital rims, nasal root, ears, and malar eminences can easily distinguish between synostotic and deformational plagiocephaly.3 Indications for surgery are to improve appearance and to relieve increased intracranial pressure when present.
CONCLUSION
Synostotic frontal plagiocephaly is most commonly caused by frontoparietal synostosis, but it can be caused by premature fusion of other sutures in the length of the coronal ring. Exact physical and radiographic examinations (multislice CT scan with 3D reconstruction) result in accurate diagnosis of skull abnormality and prevention of treatment delay.
REFERENCES
1. Bell WH. Modern practice in orthognatic and reconstructive surgery,Dallas: WB Saunders Co; 1992. pp. 1845–1854.
2. Francel PC, Park TS, et al. Frontal plagiocephly secondary to synop-sis of the frontosphenoidal suture. Case report. J Neurosurg 1995 Oct;83(4):733–736. DOI: 10.3171/jns.1995.83.4.0733.
3. Bruneteau RJ, Mulliken JB. Frontal plagio-cephaly: synostotic, compensational, or de-formational. Plast Reconstr Surg 1992 Jan;89(1):21–31. DOI: 10.1097/00006534-199289010-00005.
4. de Ribaupierre S, Czorny A, et al. Frontosphenoidal synostosis: a rare cause of unilateral anterior plagiocephaly. Childs Nerv Syst 2007 Dec;23(12):1431–1438. DOI: 10.1007/s00381-007-0469-4.
5. Davutoglu M, Okur N, et al. Craniosynostosis associated with lacunar skull: three-dimensional computed tomography features. Eur J Gen Med 2010;7(1):104–106. DOI: 10.29333/ejgm/82829.
6. Rogers GF, Proctor MR, et al. Uni-lateral fusion of the frontosphenoidal suture: a rare cause of synostotic frontal plagiocephaly. Plast Reconstr Surg 2002 Sep;110(4):1011–1015. DOI: 10.1097/01.PRS.0000021308.37931.18.
7. Schmidek A, Sweet WH. Operative neuro-surgical techniques, indications, methods and results,Philadelphia: WB Saunders Co; 2000. pp. 126–128.
8. Khanna P, Thapa M, et al. Pictorial essay: the many faces of craniosynostosis. Indian J Radiol 2011;21(1):1–9. DOI: 10.4103/0971-3026.76055.
9. Sun PP, Persing JA. Craniosynostosis. In: ed. AL, Albright IF, Pollack PD. Adelson ed. Principles and practice of pediatric neurosurgery. New York: Thieme Medical; 1999. pp. 219–242.
10. Aviv RI, Rodger E, et al. Craniosynostosis. Clin Radiol 2002;57:93–102. DOI: 10.1053/crad.2001.0836.
11. Ghali GE, Sinn DP, et al. Management of nonsyndromic craniosynostosis. Atlas Oral Maxillofac Surg Clin North Am 2002;10:1–41. DOI: 10.1016/S1061-3315(01)00003-8.
12. Zoller JE, Kubler AC, et al. Kraniozynostosen. In: ed. JE, Zoller AC. Kubler ed. Kraniofaziale chirurgie. Stuttgart: Thieme; 2002.Chapter 2.
13. Dundulis JA, Becker DB, et al. Coronal ring involve-ment in patients treated for unilateral coronal craniosynostosis. Plast Reconstr Surg 2004 Dec;114(7):1695–1703. DOI: 10.1097/01.PRS.0000142474.25114.CB.
14. Cohen MM.Jr The etiology of craniosynostosis. In: ed. MM. Jr Cohen ed. Craniosynostosis: Diagnosis, Evaluation and Management. New York: Raven Press; 1986. pp. 65–66.
15. Glass IA, Chapman S, et al. A distinct autosomal dominant craniosynostosis-brachydactyly syndrome. Clin Dysmorphol 1994;3:215–223. DOI: 10.1097/00019605-199407000-00005.
16. Muenke M., Gripp KW, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 1997;60:555–564.
17. Marsh JL, Gado MH, et al. Osseous anatomy of unilateral coronal synostosis. Cleft Palate J 1986;23:87–100.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.